微生物比较基因组学¶
**2024.11.9
思维导图¶
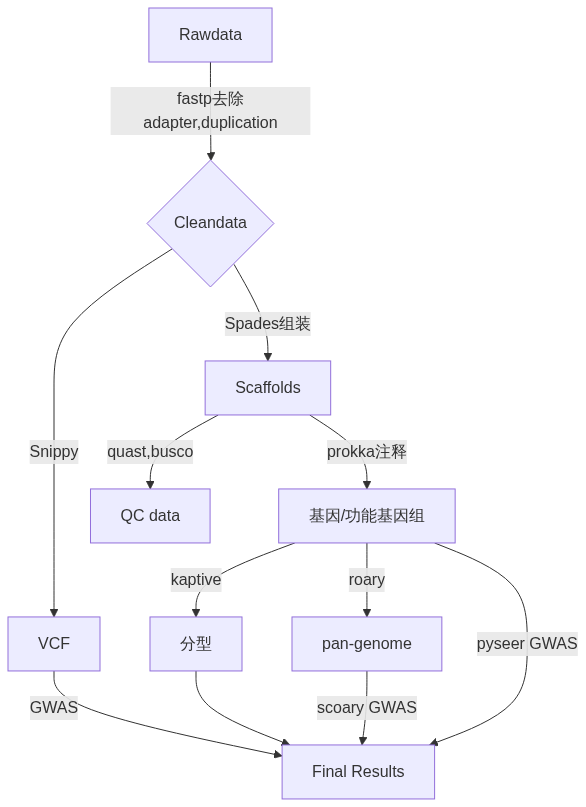
flowchart TD
A[Rawdata] --fastp去除adapter,duplication--> B{Cleandata}
B -- Snippy --> C[VCF]
B -- Spades组装 --> D[Scaffolds]
D -- quast,busco -->H[QC data]
C --GWAS--> F[Final Results]
D --prokka注释--> E[基因/功能基因组]
E --kaptive--> G[分型]
E --roary--> I[pan-genome]
I --scoary GWAS-->F
E --pyseer GWAS-->F
G -->F[Final Results]
[
质控QC¶
- fastp(去除低质量序列,去除重复序列)
- Multiqc (整合多个质控结果)
第一次质控¶
# -i 输入序列R1
# -I 输入序列R2
# -o 输出序列R1
# -O 输出序列R2
# --dedup 进行去重复操作
# -w 8 线程数
# -j 输出json格式的报告
# -h 输出网页格式的报告
fastp -i L1EHH1600255--RJ1450.R1.raw.fastq.gz -I L1EHH1600255--RJ1450.R2.raw.fastq.gz -o ./cleandata/L1EHH1600255--RJ1450.R1.clean.fastq.gz -O ./cleandata/L1EHH1600255--RJ1450.R2.clean.fastq.gz --dedup -w 8 -j ./cleandata/L1EHH1600255--RJ1450.fastp.json -h ./cleandata/L1EHH1600255--RJ1450.fastp.html
# 写一个循环批量运行
while read id
do
echo "fastp -i ${id}.R1.raw.fastq.gz -I ${id}.R2.raw.fastq.gz -o ./cleandata/${id}.R1.clean.fastq.gz -O ./cleandata/${id}.R2.clean.fastq.gz --dedup -w 8 -j ./cleandata/${id}.fastp.json -h ./cleandata/${id}.fastp.html "
done<id.txt > 01.fastp.sh
nohup bash 01.fastp.sh &
整合第一次质控结果¶
multiqc ./
第二次质控¶
fastp -i L1EHH1600255--RJ1450.R1.clean.fastq.gz -I L1EHH1600255--RJ1450.R2.clean.fastq.gz -Q -h ./cleandata2/L1EHH1600255--RJ1450.fastp.html -j ./cleandata2/L1EHH1600255--RJ1450.fastp.json -w 8
# 写一个循环批量运行
while read id
do
echo "fastp -i ${id}.R1.clean.fastq.gz -I ${id}.R2.clean.fastq.gz -Q -w 8 -j ./cleandata2/${id}.fastp.json -h ./cleandata2/${id}.fastp.html "
done<id.txt > 02.fastp.stats.sh
nohup bash 02.fastp.stats.sh &
整合第二次质控结果¶
multiqc ./
组装Assembly¶
# --careful 仔细地组装
# -1 质控后的R1
# -2 质控后的R2
# -t 12 线程数为12
# -o 输出目录(指定文件夹名称)
spades.py --careful -1 L1EHH1600255--RJ1450.R1.clean.fastq.gz -2 L1EHH1600255--RJ1450.R2.clean.fastq.gz -t 12 -o 03assembly/RJ1450
# 写一个循环批量运行
while read id1 id2
do
echo "spades.py --careful -1 ${id1}.R1.clean.fastq.gz -2 ${id1}.R2.clean.fastq.gz -t 12 -o 03assembly/${id2}"
done<id12.txt > 03.spades.sh
nohup bash 03.spades.sh&
## !!组装后记得改文件名!!
find -maxdepth 2 -name scaffolds.fasta > id1.txt #找到文件路径
cat id1.txt |cut -d "/" -f 2 > id2.txt #提取文件名
paste id1.txt id2.txt > id12.txt #生成文件路径和文件名的对应表格
while read id1 id2; do cp ${id1} ./${id2}/${id2}.fna; done<id12.txt #生成新的文件
#seqkit replace -p .+ -r "scaffold_{nr}" RJ1450.fna > RJ1450.newname.fasta
while read id; do echo "seqkit replace -p .+ -r '${id}_scaffold_{nr}' ${id}.fasta > ${id}.fna"; done<id2.txt #修改文件内容
组装后的质控¶
-
CheckM 用来评估完整性和污染度
-
Quast 评估基因组组装的连续性和一致性
-
Busco 评估基因组中功能性基因的保守性和完整性
singularity exec ~/download/quast_5.0.2.sif quast ./assmebly/* -r ~/download/ref.fna #会自动生成文件夹
注释Annotation¶
- prokka
singularity exec ~/download/prokka.sif prokka --outdir RJ1450 --prefix RJ1450 --kingdom Bacteria --cpus 8 --locustag RJ1450 --force --evalue 0.001 --proteins ~/download/kp.faa RJ1450.fna
# 自己写一个循环